ТИРОНЕТ – все о щитовидной железе Для специалистов Журнал Тиронет Архив журнала 2001 год № 3
Резистентность к тиреоидным гормонам
О. Баккер, В.М. Версинга Амстердам (Нидерланды)
Division of Endocrinology, Academic Medical Centre, Meibergdreef 9, 1105 AZ Amsterdam,
The Netherlands Thyroid international 3 - 2001
Русский перевод к.м.н. Фадеева В.В. (примечания переводчика отмечены *)
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
У первого пациента с синдромом RTH, который был описан в 1967 году, как указывалось выше, имели место яркие клинические симптомы заболевания. В данном случае речь шла об аутосомно-рецессивном варианте наследования RTH. После того как была выявлена тесная ассоциация синдрома RTH с локусом TRb [Usala, 1988], было описано около 600 случаев этого синдрома. У первого пациента была выявлена гомозиготная делеция аллеля TRb1, что, по сути, оказалось исключением из правил, поскольку у всех остальных пациентов обнаруживались точковые мутации и короткие делеции гена TRb1. В результате мутаций гена TRb1 происходит изменение последовательности аминокислот рецепторного белка или полностью прекращается его синтез. Интересен тот факт, что мутации, обнаруженные к настоящему времени в этом регионе, локализуются в трех областях, наиболее склонных к мутациям (рис. 5) [Collingwood, 1998].

Мутации, которые были обнаружены в TRb, расположены в трех областях лигандсвязывающего
домена, которые разделены областями, ответственны-ми за связывание корепрессора
и димеризацию. Аминокислотные позиции, на протяжении которых были обнаружены мутации,
обозначены над класте-рами.
В областях рецептора, которые ответственны за связывание корепрессора и димеризацию, мутаций не обнаружено, что свидетельствует о значимости этих участков для функционирования рецептора.
Наследование точковых мутаций происходит аутосомно-доминантно, при этом у пациентов выявлялось их гетерозиготное носительство. В 15% случаев RTH речь идет о спорадических случаях, то есть мутации развиваются «de novo».
Каким же образом мутация в гене рецептора приводит к формированию RTH? При анализе первого случая становится понятным, что потеря целого гена может привести к развитию RHT только в случае потери обоих аллелей. В остальных случаях, когда речь идет об аутосомно-доминатном наследовании и гетерозиготном носительстве гена, оказывается, что функция поврежденного гена не компенсируется другим нормальным аллелем.
По результатам экспериментов in vitro выяснилось, что в данном случае, мутировавший аллель оказывает негативно-доминантное действие, то есть обуславливает снижение эффектов тиреоидных гормонов даже в присутствии нормального аллеля [Sakurai, 1990; Collingwood, 1994]. Для того, чтобы мутировавший рецептор оказал негативно-доминантное действие, необходимо его связывание с ДНК и образование димера [Nagaya, 1993]. Когда в результате мутации нарушается хотя бы одни из этих процессов, негативно-доминантный эффект сказываться не будет. Исходя из имеющихся представлений о нормальной работе TR, можно предположить несколько возможных вариантов нарушения его функционирования в результате мутации. (рис. 6). Как было показано на рис. 5, все известные мутации RTb расположен в трех кластерах. В трехмерной пространственной структуре TR эти кластеры располагаются рядом с участками, ответственными за связывание тиреоидных гормонов (рис. 7). Таким образом, первым возможным вариантом RTH является потеря способности связывать тиреоидные гормоны. В этом случае гормон не может связаться с рецептором, который в дальнейшем оказывается неспособным отсоединить корепрессор (рис. 6). Другие мутации могут приводить к тому, что рецептор может связать Т3, но отсоединяет корепрессор медленнее, чем нормальный [Safer, 1998; Clifton-Bligh, 1998].
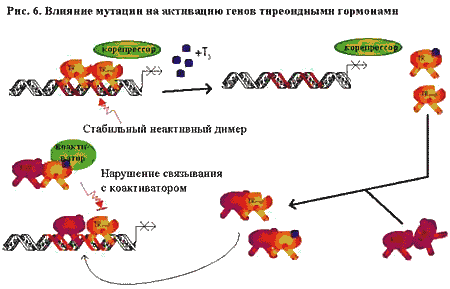
Измененный в результате мутации TR еще может связываться с TRE в виде гомодимера
с нормальным TR. Этот комплекс может взаимодействовать с корепрессором, но поскольку
с ним не может соединиться Т3, он остается стойким и неактивным. В случае, когда
у комплекса сохраняется какая-то остаточная Т3-связывающая активность, гомодимер
может отсоединиться, но измененный мутацией TR может преимущественно связывается
с RXR или другим кофактором. В этом случае, когда измененный рецептор может образовывать
гетеродимер, а в дальнейшем связываться с TRE, он блокирует доступ коактиватора
к гетеродимеру неизмененного рецептора.
Еще одно возможное влияние мутации заключается в том, что молекула мутировавшего рецептора образуют гетеродимеры, которые, не распадаясь, связываются с TRE и оказываются неспособными соединиться с коактиватором, в результате чего, не происходит активация транскрипции генов.
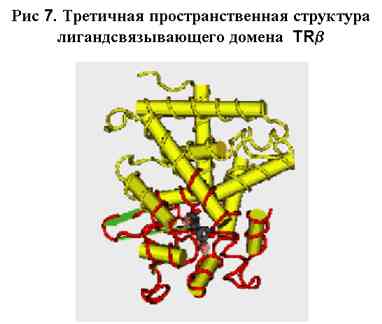
Трехмерная структура лигандсвязывающего домена TRb показана лентой, расположенной
на базовой белковой структуре [воспроизведено из базы данных NCBI-MMDB, структура
1BSX]. Верхняя часть этой структуры представляет собой ДНК-связывающий домен.
Красной лентой показаны участки, где чаще всего возникают мутации; они сконцентрированы
вокруг Т3-связывающей области.
При разрушении гена TR у мышей, моделируется фенотип, похожий на первый из описанных типов RTH, который развивается при гомозиготном наследовании делеции [Forrest, 1996]. У этих животных выявляются тяжелые слуховые нарушения. Как уже указывалось, если эта делеция наследуется гетерозиготно, все тесты, исследующие функцию щитовидной железы оказываются в норме. Экспрессия мутантного гена TR у мышей приводит к их более низкому весу, гиперактивности и снижению способности к обучению, то есть к изменениям, аналогичным таковым у пациентов с RTH [Zhu, 1999; Wong, 1997; McDonald, 1998; Abel, 1999].
Другой интересный феномен заключается в том, что у одного и того же пациента могут сосуществовать совершенно различные симптомы со стороны периферических тканей. Молекулярно-генетические исследования не обнаружили генетических различий между генерализованной и периферической формами RTH. Одни и те же мутации, которые приводят к развитию периферической RTH в одной семьи, могут обусловить генерализованную RTH в другой. Есть даже сообщения, указывающие на то, что эти две формы RTH могут выявляться в одной и той же семье и быть связанными с одной и той же мутацией. Обсуждается вопрос о том, что различия между этими формами в определенной мере субъективны и базируются на незначительной выраженности тех или иных симптомов в отдельных случаях. Генерализованную и периферическую RTH можно рассматривать как две разные формы одного и того же генетического дефекта. Возможной причиной клинической гетерогенности RTH, может быть тот факт, что у разных пациентов индивидуально варьирует уровень экспресии TR (нормальных и измененных) в периферических тканях [Mixson, 1993]. Преимущественная экспрессия тех или иных рецепторов может приводить к формированию различных фенотипов (см. рис. 6). Кроме того, не все мутации имеют одинаковое влияние на связывание рецептора с Т3 [Hayashi, 1995]. Недавно было выявлена зависимость между степенью нарушения связывания Т3 и результатами тестов, исследующих функцию щитовидной железы [Ercan-Fang, 2000]. Еще одним фактором, обуславливающим фенотипическую гетерогенность, может быть различающееся распределение TR в различных тканях (см. рис. 2). Например, в сердце преимущественно экспрессируется TRa. Если у пациента с неизмененным TRa вследствие RTH имеет место высокий уровень циркулирующего Т3, можно предположить, что у него будет выражена тахикардия, то есть один из наиболее важных симптомов тиреотоксикоза.
Продолжая обсуждать факторы, которые могут обусловливать клиническую гетерогенность RTH, следует указать на то, что у разных людей в отдельных тканях может варьировать экспрессия как TR, так и коррепрессоров и коактиваторов. В этой связи, можно указать на интересное наблюдение, в соответствии с которым мутации TRb2 проявляются клинически более выраженным фенотипом, по сравнению с мутациями TRb1. Поскольку TRb2 слабо экспрессируется в гипофизе, мутации в этом гене вероятно обуславливают периферическую форму RTH.
Недавно было описано несколько случаев, при которых несмотря на характерные для RTH гормональные и другие лабораторные сдвиги, не было выявлено мутации в гене TR [Pohlenz, 1999]. Этот феномен был объяснен отсутствием в тканях кофакторов [Weiss 1993; Weiss 1996]. Последняя концепция подтверждается результатами недавних исследований, обнаруживших у мышей, лишенных ретиноевого Х рецептора (RXR)g, симптомы, аналогичные периферическому варианту RTH [Brown, 2000]. Похожие данные были получены при исследовании мышей, лишенных коактиватора стероидного рецептора (SRC)-1 [Weiss, 1999b].
Быстрое развитие наших представлений о структуре рецепторов тиреоидных гормонов, позволяет предполагать, что коррекция тех или иных рецепторных дефектов в перспективе может осуществляться назначением их агонистов или антагонистов. Так, например, было показано, что одна из мутаций, приведшая к развитию RTH, обусловливала сдвиг лигандсвязывающего домена на 0,3 Å, что приводило к нарушению связывания с Т3. Если бы был обнаружен агонист рецептора, который мог бы с ним взаимодействовать несмотря на это небольшое изменение структуры домена, его введение могло бы корригировать RTH.




